
Заседание секции посвящено докладам студентов и аспирантов, использующих в своих исследованиях методы классической и квантовой молекулярной динамики, метод Монте Карло, методы ab initio расчетов электронной структуры материалов, многомасштабные методы, основанные на атомистическом уровне описания вещества.
Формат проведения: очно-дистанционный
Дата и место проведения: 06 апреля 2023г. в 11:00 часов, МФТИ, Синий читальный зал главного корпуса
В работе изучаются особенности Si-Al наногубок, для которых в настоящее время нет экспериментальных данных. Вычислены пороговые скорости охлаждения для наночастиц различного состава, предложен критерий, согласно которому её можно определять. Исследовано влияние параметров эксперимента на допустимые размеры наногубок, улучшена теория по их описанию, а также проанализирована её чувствительность. Приведены геометрические параметры кристаллических подсистем и их атомные составы.

В данной работе был разработан метод расчета температуры плавления материалов с систематически улучшаемой точностью. В основе метода лежит байесовский алгоритм анализа данных моделирования сосуществования в NPT ансамбле.
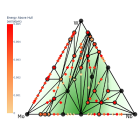
В рамках исследования был разработан высокопроизводительный алгоритм построения выпуклых оболочек (диаграмм стабильности) для сплавов металлов с заданным типом кристаллической решётки. С помощью него был проведен поиск новых сплавов с ОЦК решёткой на основе тугоплавких металлов (V, Nb, Mo, Ta, W), а также сплавов благородных металлов (Pd, Ag, Pt, Au) с ГЦК решеткой.
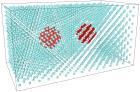
Настоящая работа посвящена исследованию взаимодействия между твердыми наночастицами в жидкой среде, возникающего на малых расстояниях при перекрывании межфазных областей. Исследование выполнено методом молекулярной динамики, в котором моделирование осуществляется на уровне атомов путем решения уравнений движения. Для описания межатомного взаимодействия использовался потенциал Леннарда-Джонса, причем для различных пар атомов задавались различные параметры потенциала.

В рамках проведенного исследования была получена аппроксимация низшей поверхности потенциальной энергии (ППЭ) $$^3A^{\prime\prime}$$ системы $$\text{O}(^3P)+\text{H}_2$$ на основе данных, полученных методами ab initio, были проведены расчеты поуровневых констант скорости процессов в данной системе с использованием полученной аппроксимации ППЭ и было проведено сравнение полученных констант с аналогичными оценками в рамках феноменологических моделей.

В пространственно-однородной кристаллической системе присутствует наивысшая частота колебаний частиц, ограничивающая спектр их колебаний. За счет однородности таких систем эта наивысшая частота не зависит от выбора частиц в структуре. При этом встречаются примеры упорядоченных систем, например, система экранированных кулоновских зарядов в поле ловушки, которые обладают пространственной неоднородностью структурных свойств. Их колебательные свойства являются объектом исследования в данной работе.

В данной работе исследуется зависимость модуля Юнга и предела упругости от пространственного распределения пор вдоль направления прикладываемой нагрузки при сжатии. Сравнивались два случая: случайное распределение пор и равномерное распределение. Исследовались модели с пористостью 22.5%, 38.5% и 58%.

В данной работе были проведены расчеты в рамках теории функционала электронной плотности в большом каноническом ансамбле, что позволило моделировать систему при заданном электрохимическом потенциале. Применение данной методики позволило рассчиать теормодинамиру рекции восстановления кислорода на N-допированных графенах при фиксированном потенциале электрода. Показана каталитическая активность трех рассмотренных дефектов графена, содержащих атом азота, по отношению к РВК.

В данной работе методом молекулярной динамики изучено влияние атомов водорода, накопленных на θ′-фазах, на деформационное поведение сплава Al-Cu при сдвиге и растяжении. Исследовано влияние размера, температуры и скорости на развитие деформаций в рассматриваемых системах. В работе мы рассмотрели системы, содержащие включения двух типов θ′.

В настоящей работе при помощи теории функционала электронной плотности (DFT) изучаются причины повышенной дестабилизации обратным α-эффектом циклических пероксикарбениевых ионов. Полученные результаты раскрывают роль данного эффекта в каскаде образования трипероксида ацетона.

В данной работе были изучены дислокации, образующиеся в мононитриде урана, при различных ориентациях атомных плоскостей в расчетной ячейке. Расчеты проводились методом молекулярно-динамического моделирования с использованием программного пакета LAMMPS. При расчетах были использованы различные межатомные потенциалы и изучалась зависимость образующихся дислокаций от используемого потенциала.
Проводится моделирование решётки урана с растворёнными в ней атомами ксенона. Анализируются пузырьки ксенона, которые образуются в результате этого

В настоящей работе мы рассматриваем разработку программного кода, позволяющего использовать результаты расчета электронного спектра в Quantum Espresso для расчета эффективной массы и термоэлектрических свойств в AICON2. На примере соединения SnSe сравниваются результаты расчетов эффективной массы на основе электронной структуры, полученной в VASP и Quantum Espresso.
В данной работе мы применяем магнитный многокомпонентный машинно-обучаемый потенциал mMTP для исследования системы Fe-Al при различных концентрациях алюминия и железа. В ходе исследования были рассчитаны энергии образования структур, равновесные параметры решётки и магнитные моменты ячейки. Показано, что рассчитанные с помощью многокомпонентного mMTP потенциала величины близки к аналогичным величинам, полученным при помощи первопринципных методов.

В данной работе мы изучили процесс нуклеации алмаза в графите начиная с формирования сверхтонких алмазных слоёв. Далее мы использовали методы машинного обучения для получения потенциала, с помощью которого затем было проведено изучение роста алмаза в матрице графита. Был рассчитан термодинамический потенциал Гиббса при различных давлениях и были определены барьеры нуклеации алмаза в графите.

В данной работе разработан алгоритм моделирования процесса PECVD (Plasma-enhanced chemical vapor deposition), используемого для нанесения тонких плёнок различных материалов. Применялся метод молекулярной динамики с машинно-обучаемым потенциалом(Moment Tensor Potential) в качестве модели межатомного взаимодействия.

Расчет энергии диссоциации, равновесной структуры и колебательных частот тетраазота N4 методами квантовой химии.

В этом исследовании представлены новые сверхпроводящие гидриды в системе Ca-Y-H, теоретически предсказанные с помощью эволюционного алгоритма USPEX под высоким давлением (180 ГПа). С помощью DFPT и формализма Мигдала-Элиашберга, было изучено сверхпроводящее состояние полученных гидридов, в итоге было показано, что они обладают высокой критическом температурой сверхпроводимости (от 204 до 269 К). Термодинамическая стабильность системы была исследована с учётом вклада энергии нулевых колебаний.

В данной работе был использован программный комплекс Quantum ESPRESSO для расчёта магнитных, электронных и структурных характеристик монослоя VN в ферромагнитном состоянии. Результаты расчётов позволяют сделать вывод о свойствах исследуемого материала: о характере химических связей, энергии отслойки, стабильности модификаций.

Создание систем, улучшающих свойства лекарств и адресную доставку в органы, является важным направлением в развитии нанотехнологий. В работе рассмотрен процесс взаимодействия молекул антибиотика с потенциальным наноносителем, для этого использован метод молекулярной динамики, позволяющий исследовать процесс самосборки молекул и их взаимодействие с препаратом на молекулярном уровне.

Одной из актуальных задач современного материаловедения является поиск функциональных материалов с уникальными свойствами, такими как двулучепреломление и генерация второй гармоники. В последнее время подходы, связанные с методами машинного обучения, стали все чаще использоваться для поиска материалов с заданными свойствами. В нашей работе рассматривается применение графовой нейронной сети ALIGNN для определения оптических коэффициентов Δn и SHG различных оптических кристаллов.
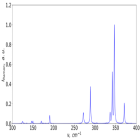
Слоистые и двумерные материалы в последнее время привлекают огромный интерес благодаря необычным оптическим свойствам, одно из которых – оптическая анизотропия. As2S3 имеет слоистую структуру и уникален наличием внутриплоскостной анизотропии, что делает исследование его кристаллической структуры и связанных с ней свойств интересной и важной задачей. В данной работе с помощью первопринципных методов был рассчитан рамановский спектр сульфида мышьяка (III) (P21/n).

В работе представлены результаты компьютерного моделирования соли Фриделя (Cl-гидрокалюмит,Ca2[Al(OH)6]· Cl·2H2O), с помощью методов ab initio молекулярной динамики (МД), а также классической МД. Проведен анализ фазового перехода, обноруженного эксперементально при температуре ~308K. Исследованы кристаллографические, структурные и динамические параметры вещества в различных фазовых состояниях.
Недавно обнаруженные металлоценоподобные структуры MCp2, представляющие собой атомы переходных металлов между двумя листами графенового аллотропа (Octite M1 или New 17), способны обладать дальним магнитным порядком. В рамках теории функционала электронной плотности и приближения DFT+U были исследованы магнитные свойства стабильных структур t_MCp2 и m_MCp2 (M = Co, V, Cr): найдены обменные константы и энергии магнитной анизотропии.

Данная работа посвящена разработке межатомного потенциала для описания взаимодействия в системе свинец-кислород. Целью создания такой модели является описание физико-химических процессов, протекающих в объеме свинцового расплава с участием кислорода, при помощи методов атомистического моделирования.
В данной работе исследованы механические и термодинамические свойства монослоя NiCl2 с применением теории функционала плотности и метода молекулярной динамики. Для использования последнего необходимо знать эмпирический потенциал, который отсутствует в литературе для данного материала. Значения параметров эмпирического потенциала были рассчитаны при помощи алгоритма оптимизации Нельдера-Мида.