
На секцию будут приниматься доклады по исследованию электронной структуры, потенциальных поверхностей, спектральных параметров, термохимических данных, реакционной способности и динамике превращений молекул. Работы по разработке новых методов, алгоритмов и программ расчета волновых функций, строения, свойств и превращений молекул на основе представлений квантовой теории и методов вычислительной математики.
Формат проведения: очно-дистанционный
Дата и место проведения: 05 апреля 2023г. в 10:00 часов, МФТИ, лабораторный корпус, 505 ЛК

Релятивистским методом симметризованных линеаризованных присоединенных цилиндрических волн рассчитано влияние торсионных деформаций на электронные и спиновые свойства нехиральных (7, 7) кремниевых и углеродных нанотрубок. Деформация кручения снимает спиновое вырождение и приводит к образованию щелей на краях зон. Она работает как переключатель: противоположные направления скручивания приводят к противоположной хиральности нанотрубки и индуцируют противоположные спиновые расщепления и токи.
В работе представлен теоретический метод для моделирования спектров однофотонного и двухфотонного поглощения флуоресцентных меток и зондов в реальном окружении с учетом однородного и неоднородного уширения. Использование красителя Fura-2 при двухфотонном возбуждении решает проблему фототоксичности и фотообесцвечивания, а так же позволяет применять долговременное непрерывное линейное сканирование для отслеживания изменения концентрации ионов кальция во многих типах клеток.

Рассмотрены механизмы химических реакций CH3CHO+NН3→CH3CH(OH)NH2→CH3CHNH+H2O и CH3CH(OH)NH2+CH3CHO→[CH3CH(OH)]2NH→CH3CH(OH)NCHCH3+H2O во льдах аммиака путем квантово-химических вычислений. Условия твердой фазы были созданы с помощью подхода SCRF/SMD. Построены диаграммы поверхности потенциальной энергии химических реакций.

Предложена методология для расчета сечения двухфотонного поглощения белка EGFP и его химических модификаций методами молекулярной динамики и квантовой химии. Представлена N-уровневая модель, позволяющая рассчитывать компоненты тензора двухфотонного поглощения. Исследована зависимость сечения от конформации активного центра модификации EGFP T203I. Показано влияние мутации T203I на сечение двухфотонного поглощения белка EGFP и предложен способ увеличения сечения путем введения точечных мутаций.

Построена поверхность потенциальной энергии химической реакции пропионитрила с метиновым радикалом с использованием методов квантовой химии и статистической физики. Оптимизированы геометрии структур на поверхности потенциальной энергии методом ꞷB97xd с базисным набором cc-pvtz. Относительные энергии всех структур рассчитаны с применением явно коррелированной теории связанных кластеров CCSD(T)-F12 и корреляционно-согласованного базисного набора cc-pvtz-f12.

В работе разработан и реализован метод расчета фотоэлектронных спектров и спектров фотоионизации при колебательной автоэмиссии из слабосвязанных электронно-возбужденных состояний молекулярных анионов. Метод основан на расчете матричных элементов неадиабатического взаимодействия в пространстве как электронных, так и ядерных переменных в приближении Кондона с учетом эффекта Душинского.

В работе продемонстрирована методика моделирования фотоэлектронных спектров анионных биохромофоров в растворе при двухфотонном резонансном возбуждении на примере фенолят-иона – типичного фрагмента многих хромофоров флуоресцентных белков.
Полученные результаты позволяют интерпретировать экспериментальные фотоэлектронные спектры фенолят-аниона в водном растворе при резонансном двухфотонном возбуждении в УФ диапазоне.

Ab initio исследование реакции радикалов бензила и фенила, целью которого является построение поверхности потенциальной энергии данной реакции. Основным её продуктом является флуорен — простейший полициклический ароматический углеводород, содержащий пятичленное кольцо.
Вычисления энергий реагентов, продуктов, промежуточных и переходных состояний проводились на уровне теории G3(MP2, CC)//B3LYP, геометрии и энергии нулевых колебаний посчитаны методом B3LYP/6-311G**.
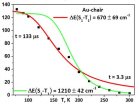
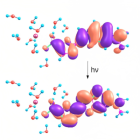
В данной работе методом TD-DFT были рассчитаны электронные спектры поглощения ряда диядерных комплексов монетных металов, которые находятся в прекрасном согласии с экспериментом, а также определена природа координационных связей, оценина их энергия по орбитальному вкладу (метод DFT), исследованы кинетики люминисценции по моделе TADF. Изучаемые в работе соединения являются эффективными и высокостабильными металл-органическими люминофорами, применимые в технологии OLED.

В настоящей работе представлен метод вычисления многоцентровых молекулярных интегралов от оператора обобщённого релятивистского псевдопотенциала и его программная реализация – библиотека подпрограмм LIBGRPP. Отклонения рассчитанных в рамках модели GRPP вертикальных энергий возбуждения молекул ThO и UO2 от полноэлектронного результата (гамильтониан Дирака-Кулона-Гонта) не превышают 50 см-1 для 31 состояния ThO и 110 см-1 для 110 состояний UO2.

Представлен алгоритм ранжирования физических параметров в зависимости от их влияния на кинетику данной молекулы в процессе химических реакций в молекулярном облаке. С помощью математической статистики и машинного обучения "c учителем" удалось оценить важность каждого из заданных параметров для каждой молекулы, присутствующей в сетке реакций.

Рассмотрена задача вычисления энергии связывания пары «лиганд-белок» (в заданных активных центрах) в растворе для белков PCAF и NRAM и лекарственных соединений из базы данных ChEMBL. Проведено сравнение моделей PM7, SE/MM (PM7/Amber), Vinardo.