
Темы работ, которые рассматриваются на секции:
- Классическая молекулярная динамика;
- Квантовое атомистическое моделирование;
-Методы Монте-Карло в статистической термодинамике и физической кинетике
- Технологии суперкомпьютерного моделирования;
-Многомасштабные подходы, основанные на атомистическом моделировании.
Формат проведения: очный
Рабочий язык: русский
Дата и место проведения: 07 - 08 апреля в 11:00 часов, 7 апреля - синий читальный зал главного корпуса МФТИ, 8 апреля - МФТИ, биологический корпус, 107БК

В работе решается задача поиска зависимости между пределом длительной прочности конструкционных материалов и их составом. Для этого была собрана и проанализирована база данных по соответствующим экспериментам. Затем исследованы важные зависимости и с их помощью упрощена задача. Далее были построены предсказательные модели и вычислена их точность. С помощью моделей был произведён поиск составов кандидатных материалов для заданных параметров предела длительной прочности.
Расчёт сечения фотоионизации фотовозбуждения для плазмы гелия по методу Берджесс-Ситона. Сравнение функции сечения от энергии фотона в припороговой области с плотностью сил осцилляторов.

Переход плотного нагретого флюидного водорода из состояния изолятора в проводящее состояние при давлениях порядка 20-400 ГПа и температурах 500-5000 К является предметом активных научных исследований в течение последних нескольких десятилетий. В данной работе предлагается новый механизм экситонной диссоциации, способеный дать количественное описание множества экспериментальных результатов, а также устранить расхождения между наблюдениями различных групп.

Целью настоящей работы является создание математической модели для оценки влияния отдельных аминокислот в ТМ сегментах РТК и искусственных полипептидах на пространственное распределение связанных липидов вблизи мономерных ТМ спиралей белков при помощи методов молекулярного моделирования. В ходе работы были разработаны методы изменения и анализа формы поверхности пептидов. На примере аминокислотных остатков аланина и глицина было показано влияние формы на поведение липидного бислоя.
В данной работе исследуется структура водного раствора сахарозы методами атомистического моделирования. Мы обнаружили несколько устойчивых конформаций молекулы сахарозы, стабилизируемых внутримолекулярными водородными связями, и рассчитали их времена жизни.
В данной работе получены возможные гетеродимерные состояния ТМ доменов DDR1 и ErbB2, количественно оценена склонность рассматриваемых пептидов к ассоциации в липидном бислое. Выявлены взаимодействия, стабилизирующие предполагаемые структуры гетеродимеров, и предложены потенциальные способы воздействия на исследуемые белки.

В работе предложены и исследованы некоторые методы расчета траекторий системы атомов с использованием моделей машинного обучения. Подобный подход может позволить значительно ускорить вычисление траекторий атомов для систем со сложной функцией потенциала. Также данный метод можно использовать для получения траекторий системы с неизвестным априори потенциалом, а известными лишь небольшими частями траекторий, входящих в нее компонентов.

Рассчитывается коэффициент диффузии для монокристалла чистого диоксида циркония и легированного иттрием. Рассматривается модель без учета поляризации и с учетом поляризации.
В данном исследовании рассматривается точность воспроизведения коэффициента поверхностного натяжения н-нонана в рамках модели SAFT-ɣ Mie. Для этого была получена зависимость коэффициента поверхностного натяжения от температуры и было рассчитано значение парахора модели SAFT-ɣ Mie н-нонана.
В данной работе проводится сравнительная оценка межатомных потенциалов GAFF, OPLS-AA/CM1A, CHARMM36 и COMPASS по точности расчета плотности и вязкости диизопропилового эфира методом классической молекулярной динамики.

В работе представлены результаты исследования динамического поведения фермент-субстратных комплексов металло-β-лактамаз L1 и NDM-1 с субстратами нитроцефином и имипенемом. Исследовалось влияние структурной подвижности активного центра фермента и строения субстратов на эффективность активации субстрата ферментом. Показано, что более структурно жесткий активный центр позволяет более эффективно активировать субстрат, что приводит к понижению барьера стадии нуклеофильной атаки.

В работе проведено молекулярное моделирование механизма ингибирования пенициллин-связывающих белков 2 (ПСБ2) из трех штаммов FA19, 35/02 и H041 бактерии Neisseria Gonorrhoeae антибиотиком цефтриаксоном. Исследование динамического поведения комплексов и несвязанных ферментов показало, что с ростом резистентности ПСБ2, доля реакционных структур фермент-субстратного комплекса падает, молекулярный механизм в активном центре изменяется, а сродство ПСБ2 к цефтриаксону понижается.

В работе рассматривается задача верификации известных силовых полей на предмет корректного воспроизведения физических свойств тройной системы полиэтиленоксид-вода-ионы. Взаимодействие полиэтиленоксид-вода проверяется на примере водного раствора диоксана [Bakulin I. et al. // J. Chem. Phys. 2021. V. 155]. Взаимодействие полиэтиленоксид-ионы проверяется на примере реакции образования комплекса молекулой 18-краун-6 эфира с ионами металлов в воде [Bakulin I.K. et al. // JPCS. 2021. V. 1787].

В работе изучаются структурные и динамические свойства цепочек пылевых частиц в потоке плазмы газового разряда путем численного молекулярно-динамического моделирования.
Стабильность водных протонных катионов и эффективность их конверсии друг в друга является важнейшим фактором протонной мобильности. Информация о доминирующем типа водного протонного катиона носит большое значение для исследования фундаментальных свойств биологических протон-транспортных цепей, отвечающих за синтез АТФ. Для моделирования образования водных катионов в жидкой воде и соляной кислоте нами был применён метод молекулярной динамики на интегралах по траекториям.

Для исследования свойств состояния с антиферромагнитным (несоизмеримым) магнитным порядком системы коллективизированных электронов, описываемых однозонной моделью Хаббарда в режиме сильных корреляций, авторами данной работы было реализовано диаграммное расширение динамической теории среднего поля (DMFT).

В данной работе проводится атомистическое моделирование керогенов ,основной составляющей сланца, а также исследуются различные упругие свойства незрелых (IA) и перегретых (IID) керогенов, в частности объемный модуль упругости и модуль Юнга. Также рассматривается взаимосвязь этих параметров с пористостью структуры.

Калиевые ионные каналы hERG и EAG являются гомологичными. Для hERG описано достаточно большое число лигандов, включая природные, а вот для EAG высокоаффинных и селективных лигандов не известно. EAG является мишенью разработки противоопухолевых препаратов. Учитывая гомологию каналов hERG и EAG и высокое сходство их пространственной структуры, мы задались целью модифицировать блокатор hERG, токсин скорпиона BeKm-1, так, чтобы он воздействовал на EAG.

Методом молекулярной динамики исследуются фазовые переходы жидкость-
кристалл в неограниченной среде и в шаровом кластере на примере потен-
циалов Леннарда-Джонса и EAM. Изучается свойства четырех критериев:
параметра Линдемана, параметра IDF, пространственного четырехточеч-
ного коррелятора и К-энтропии.

В данной работе изучается поведение локальных магнитных моментов в сильно коррелированной модели Хаббарда в рамках динамической теории среднего поля. Исследовано влияние формирования и экранирования локальных магнитных моментов на температурные зависимости и U-зависимости (кулоновское взаимодействие) функций отклика / функций восприимчивости, корреляционных функций, спектральных функций и удельного сопротивления в окрестности перехода металл-изолятор в допированном и недопированном случаях.
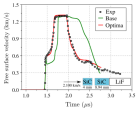
В данной работе предлагается возможный подход к решению задачи подбора параметров модели распространения ударных волн в керамиках: SiC, BC и AlN. Данный подход может быть применим практически без изменений и в других областях знаний, где стоит задача подбора параметров сложной функции (в самом общем смысле) по некоторому условию.

С помощью метода молекулярно-динамического моделирования исследуется потенциал средней силы на границе двух растворов: ионов карбоксицеллюлозы с Li+ и Li+ с Cl-

На макроскопическом уровне поведение жидкостей и газов хорошо описывается уравнениями Навье-Стокса. Долгое время исследование поведения жидкостей и газов с помощью метода молекулярной динамики было невозможно из-за недостатка вычислительных мощностей.
В данной работе представлен и реализован новый метод создания стационарного потока вещества в задаче молекулярной динамики, приведены примеры его применения и реализованы методы высокопроизводительного анализа полученных с его помощью данных.

Разработан и реализован метод анализа молекулярной коммуникации с использованием данных МД и теории информации.
В данной работе будут представлены результаты по определению энергии и энтропии образования точечных дефектов с помощью как прямого установления равновесия в молекулярной динамике, так и полностью равновесных расчётов с использованием статических расчётов, термодинамического интегрирования и метода вставок Видома.

В рамках классической молекулярной динамики рассматиравются неравновесные переходы между аморфными льдами низкой и высокой плотности. Предложен алгоритм классификации локальной структуры аморфного льда, который позволяет визуальзировать образование и дальнейший рост кластеров новой фазы в рассматриваемых переходах, а также оценить размер критического "зародыша новой фазы". Результаты сопоставляются с экспериментами и общим современным пониманием аморфно-аморфных переходов в воде.

В работе изучается применимость метода потенциала средней силы для получения свойств двухфазных бинарных систем углеводородов. Рассматривается применение метода ПСС для расчета свойств смесей, таких как свободная энергия сольвации, растворимость и константы Генри.
Вычисляются и сравниваются энергии миграции точечных дефектов в нитриде урана двумя различными методами для двух межатомных потенциалов
Данная работа посвящена поиску стабильных сокристаллов CL-20 при стандартных условиях. Моделирование структур осуществлялось при помощи специально разработанного генератора случайных кристаллических структур с фиксированным окружением заданных структурных элементов на основе экспериментальных данных о высокоэнергетическом полиморфе α-CL-20. После релаксации в LAMMPS + ReaxFF производился отбор стабильных структур и изучение структурного разнообразия.
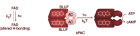
В данной работе методами молекулярной динамики и динамического сетевого анализа проводится сравнительный анализ путей фоторегуляции в бактериальных фотоактивируемых аденилатциклазах из организмов Beggiatoa и Oscillatoria acuminata.
Целью данной работы было получение и анализ корректности простой молекулярно-динамической модели дегидратированного ДГАЛ-Cl - одной из структурных модификация гидроксида алюминия, способной к сорбции лития. За основу было взято популярное силовое поле ClayFF, в котором была репараметризавались заряды алгоритмом DDEC6. Анализ корректности модели проводился на основе сравнения с экспериментальными данными и предыдущими исследованиями методом функционала электронной плотности.

В докладе будут рассмотрены теоретические основы комбинированного метода квантовой механики / молекулярной механики (КМ/ММ) для расчета энергий и сил в модельных молекулярных системах, а также метод молекулярной динамики для изучения конформационного пространства систем. Среди примеров будут рассмотрены ферментативные реакции, изучение которых имеет важное прикладное значение.